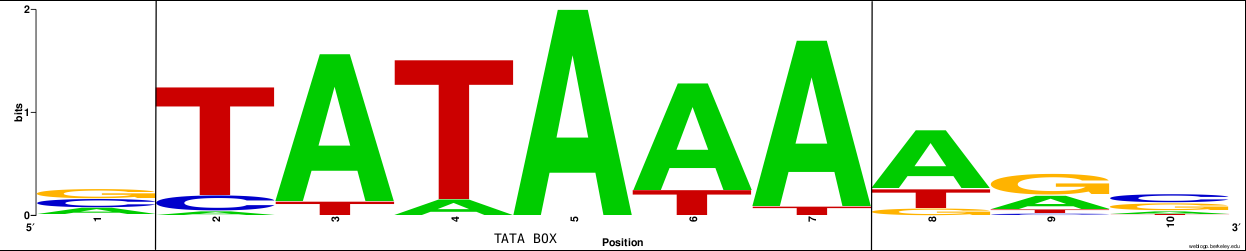
Suppose that we want to calculate the expected distance of a DNA motif within a DNA target sequence, if we know the composition bias or the probability distribution (multinomial model) we can compute it just fine.
Download the R code <- here
FIRST PART
For example, supose that we want to compute the expected distance of the motif "GATC" in a sequence composed of 10,000 bases given that the whole sequence follows a probability distribution of p(A) = 0.3, p(C) = 0.2, p(G) = 0.2, p(T) = 0.3.
So open an R prompt and load the functions:
Then, enter the initial values:
And finally, compute the average expected distance of every occurrence of the motif inside the target sequence. Using the following formula (this computation corresponds to the "computeExpectedDistance" function of the R script "motifOccurrence.R"):
expectedDistance = lengthDNAseq/(lengthDNAseq*p))
where "p" stands for the joint probability of the motif, in other words: p = p(GATC) = p(G)*p(A)*p(T)*p(C) = 0.2*0.3*0.3*0.2 = 0.0036
To easily compute the expected distance in R, type:
As you see, it returns the value of "277", this number means that, for the "GATC" motif inside a sequence of 10,000 bases with a composition bias of p(A) = 0.3, p(C) = 0.2, p(G) = 0.2, p(T) = 0.3 we may expect a distance of 277 bases between each "GATC".
Or, a little more graphic:
277 bases | GATC | 277 bases | GATC | .....
SECOND PART
Another way to compute this (even though it involves more computations) we can simulate X number of DNA sequences with a fixed length with an equal probability distribution per sequence and extract the coordinates of the motif within each sequence to finally compute the average distance of the motif.
There is a function titled "iterateComputeDistance" to do the calculations for you. Add the next parameter to the R environment:
Compute the average distance of the "GATC" motif within 100 DNA sequences (the other parameters remain equal)
As we expected, the results of the two approaches are highly similar (ouuu yeah!)
THIRD PART
But, what happens when we already have a sequence and want to know the expected distance of that motif inside of it?.
Just like "Hey dude, I have an E.coli plasmid DNA sequence and want to know the average distance of the GATC motif".
Lets test using the sequence "Escherichia coli 2078 plasmid pQNR2078 complete sequence" <- http://www.ncbi.nlm.nih.gov/nuccore/HE613857.1
Ok, use the "ape" library to import the sequence to the R environment (if this library is not installed, type: install.packages("ape"))
NOTE: the GenBank sequences are in lowecase, so it will be needed to use a motif in lowercase to do the right computations.
Import the library:
Import the sequence:
Get the coordinates:
Get the average distance between the motif occurrences.
[1] 270
Compute the number of occurrences of the motif among the plasmid (the result is 151 occurrences):
The number of occurrences of the motif in R is:
So, the main distance between the motif "gatc" finally is 270 bases and we are done :D
CODE:
Benjamin
Download the R code <- here
FIRST PART
For example, supose that we want to compute the expected distance of the motif "GATC" in a sequence composed of 10,000 bases given that the whole sequence follows a probability distribution of p(A) = 0.3, p(C) = 0.2, p(G) = 0.2, p(T) = 0.3.
So open an R prompt and load the functions:
source("motifOccurrence.R")
Then, enter the initial values:
lengthSeq <- 10000
motif <- c("G","A","T","C")
probDistr <- c(0.3,0.2,0.2,0.3)
And finally, compute the average expected distance of every occurrence of the motif inside the target sequence. Using the following formula (this computation corresponds to the "computeExpectedDistance" function of the R script "motifOccurrence.R"):
expectedDistance = lengthDNAseq/(lengthDNAseq*p))
where "p" stands for the joint probability of the motif, in other words: p = p(GATC) = p(G)*p(A)*p(T)*p(C) = 0.2*0.3*0.3*0.2 = 0.0036
To easily compute the expected distance in R, type:
expectedDist <- computeExpectedDistance(probDistr,lengthSeq,motif)
As you see, it returns the value of "277", this number means that, for the "GATC" motif inside a sequence of 10,000 bases with a composition bias of p(A) = 0.3, p(C) = 0.2, p(G) = 0.2, p(T) = 0.3 we may expect a distance of 277 bases between each "GATC".
Or, a little more graphic:
277 bases | GATC | 277 bases | GATC | .....
SECOND PART
Another way to compute this (even though it involves more computations) we can simulate X number of DNA sequences with a fixed length with an equal probability distribution per sequence and extract the coordinates of the motif within each sequence to finally compute the average distance of the motif.
There is a function titled "iterateComputeDistance" to do the calculations for you. Add the next parameter to the R environment:
iterations <- 100
Compute the average distance of the "GATC" motif within 100 DNA sequences (the other parameters remain equal)
expectedDistWithSimSeqs <- iterateComputeDistance(probDistr,lengthSeq,motif,iterations)
As we expected, the results of the two approaches are highly similar (ouuu yeah!)
THIRD PART
But, what happens when we already have a sequence and want to know the expected distance of that motif inside of it?.
Just like "Hey dude, I have an E.coli plasmid DNA sequence and want to know the average distance of the GATC motif".
Lets test using the sequence "Escherichia coli 2078 plasmid pQNR2078 complete sequence" <- http://www.ncbi.nlm.nih.gov/nuccore/HE613857.1
Ok, use the "ape" library to import the sequence to the R environment (if this library is not installed, type: install.packages("ape"))
NOTE: the GenBank sequences are in lowecase, so it will be needed to use a motif in lowercase to do the right computations.
Import the library:
require("ape")
Import the sequence:
plasmid <- read.GenBank("HE613857.1")
plasmidDNA <- as.character.DNAbin(plasmid)
plasmidDNA <- plasmidDNA[[1]]
motifEcoli <- c("g","a","t","c")
Get the coordinates:
plasmidDNAcoord <- coordMotif(plasmidDNA,motifEcoli)Get the average distance between the motif occurrences.
plasmidDNAmotifDistance <- computeDistance(plasmidDNAcoord)
[1] 270
Compute the number of occurrences of the motif among the plasmid (the result is 151 occurrences):
The number of occurrences of the motif in R is:
(length(plasmidDNAcoord)-1)
So, the main distance between the motif "gatc" finally is 270 bases and we are done :D
CODE:
#
# Script: motifOccurrence.R
# Author: Benjamin Tovar
# Date: 21/April/2012
#
################################################################################
# ############
# FUNCTIONS:
# ############
##############################################################################
iterateComputeDistance <- function(multinomialDNAmodel,
lengthDNAseq,
motif,
numberOfIterations){
# This function returns the mean distance
# of a given motif given X number of DNA sequences given a multinomial model
# (probability distribution of each base).
# So, it will generate X number of DNA sequences using a given
# probability distribution and then it will compute the distance among
# that mofit within the total set of sequences to finally returns
# the average distance of the motif.
result <- rep(NA,numberOfIterations)
for(i in 1:numberOfIterations){
currentGenome <- NA
currentCoordinatesOfMotif <- NA
currentSequence <- sample(c("A","C","G","T"),
lengthDNAseq,rep=T,
prob=multinomialDNAmodel)
currentCoordinatesOfMotif <- coordMotif(currentSequence,motif)
result[i] <- computeDistance(currentCoordinatesOfMotif)
cat(" *** Iteration number: ",i," completed *** | average distance = "
,result[i],"\n")
}
result <- trunc(mean(result))
cat(" \n*** Computation status: DONE ***\n\n")
return(result)
}
##############################################################################
coordMotif <- function(targetSequence,motif){
# This function returns the coordinates of the motif of study in a target
# DNA sequence. In other words, if I found the motif, tell me exactly in
# which position of the DNA sequence is.
lengthMotif <- length(motif)
lengthTargetSeq <- (length(targetSequence)-lengthMotif)
motif <- toString(motif)
motif <- gsub(", ","",motif)
res <- 1
for(i in 1:lengthTargetSeq){
currentTargetSeq <- targetSequence[i:(i+(lengthMotif)-1)]
currentTargetSeq <- toString(currentTargetSeq)
currentTargetSeq <- gsub(", ","",currentTargetSeq)
if(currentTargetSeq == motif){
res[(length(res)+1)] <- i
}
}
return(res)
}
##############################################################################
computeDistance <- function(coordinatesOfMotif){
# This function returns the mean distance
# of a given motif given its coordinates within a target DNA sequence.
# In other words, If I already got a list with the coordinates where the
# motif is inside a DNA sequence, tell me the average distance between
# this coordinates to get the expected distance of that motif.
currentDistance <- rep(NA,(length(coordinatesOfMotif)-1))
lengthCoord <- length(currentDistance)
for(i in 1:lengthCoord){
currentDistance[i] <- coordinatesOfMotif[i+1]-coordinatesOfMotif[i]
}
res <- trunc(mean(currentDistance))
return(res)
}
##############################################################################
computeExpectedDistance <- function(multinomialModel,
lengthDNAseq,
motif){
# This function computes the expected distance of a given motif in a DNA
# sequence given its multinomial model (probability distribution of
# each base)
# Convert the motif into an index
motifIndex <- gsub("A",1,motif); motifIndex <- gsub("C",2,motifIndex)
motifIndex <- gsub("G",3,motifIndex); motifIndex <- gsub("T",4,motifIndex)
motifIndex <- as.numeric(motifIndex)
# Compute p value of the motif given the multinomial model
p <- rep(NA,length(motif))
for(i in 1:length(motifIndex)){
p[i] <- multinomialModel[motifIndex[i]]
}
p <- prod(p)
result <- trunc(lengthDNAseq/(lengthDNAseq*p))
return(result)
}
##############################################################################
# Benjamin
Benjamin






